Elucidating the molecular basis of vision loss in Battens disease
Collaborators: Dr. Jonathan Mink, Dr. David Pearce, Dr. Jill Weimer, Dr. David Williams
Human induced pluripotent stem cell (hiPSC) technology and genome editing capabilities are scientific advances that have already, independently, revolutionized our approach to studying and treating human eye diseases. However, it is the integration of these technologies and the ability to study the disease pathophysiology and test potential drug-treatments in patient-derived cells in vitro in parallel with a patient’s own eyes in vivo which promotes the biggest advances in the field of translational vision research and therapeutics. For example, a prominent limitation of histopathologic studies in human eyes is that the postmortem retina is obtained at a late stage of the disease, making it difficult to differentiate between initial insult and secondary effects. hiPSC-based disease modeling provides an alternate platform to identify the temporal sequence of structural and pathophysiological changes that lead to disease pathology in affected patients. Furthermore, CRISPR technology and the ability to “correct” the genetic mutation in patient-hiPSCs supports the ability to define a direct correlation between the genetic aberration and the pathophysiological alteration observed in patient-derived cells. This will have significant implications for personalized clinical medicine in future years, particularly in monogenic diseases like Juvenile neuronal ceroid lipofuscinosis (JNCL).
JNCL also called Batten disease or CLN3, is a childhood-onset lysosomal storage disorder that leads to progressive neurological and visual dysfunction. In fact, vision loss and consequent blindness is hallmark of and one of the first clinical features of JNCL. It has also been well established that vision loss in JNCL is due to retinal degeneration and can mimic other more common retinopathies including Stargardt’s and cone-rod dystrophy. In fact, because vision loss in JNCL precedes other neurological abnormalities, sometimes by many years, patients with JNCL can be misdiagnosed with other common retinal degenerations. The prominence and early onset of retinal degeneration in NCLs makes it likely that cellular process(s) that are dysregulated in NCLs are critical for retinal homeostasis. Furthermore, histopathologic studies have described accumulation of autofluorescent lipopigment in retinal neurons and degeneration of multiple retinal cell layers in JNCL including the retinal pigment epithelium (RPE). However, because postmortem retina from JNCL patients is so severely degenerated, it is difficult to determine the individual involvement of specific retinal cell type(s) in the disease. The characterization of retinal changes in the early stage of the disease thus will be important for delineating the disease mechanisms Animal models of JNCL, CLN3-/- mice exist and display increased autofluorescence accumulation in the retina, decreased ERG b-wave amplitudes, reduced cone photoreceptor function, loss of INL cell density, ganglion cell death and impaired phagosome processing by RPE cell. The widespread retinal pathology affecting multiple retinal cell type(s) in CLN3-/- mice is consistent with the human disease. However, the disease in the CLN3-/- mice is delayed compared to humans with prominent phenotypic and functional alterations appearing in 12-24 month old animals. This is a huge disparity given the average life span of mice ranges between 2-3 years. Furthermore, CLN3 knockout model may not be completely representative of the disease as recent studies have suggested that the truncated CLN3 protein retains some its function. Therefore a mutation-specific human model of CLN3 would be advantageous for disease modeling studies. Human induced pluripotent stem cells (hiPSCs) offer alternate suitable platform to investigate the retina-specific disease mechanism of JNCL. Importantly, We have recently used hiPSC to create retinal models of macular degeneration (Galloway et al PNAS 2017 and Best disease (Singh et al HMG 2013) that recapitulated key features of the human disease and provided important pathophysiological insights. Building on these studies, the objective of the current project is to utilize patient-derived hiPSC model of JNCL for investigating disease mechanisms and testing drug therapies. Of note, to complement our in vitro studies in patient-derived hiPSC-retinal cells, when relevant and applicable, major findings the study will be validated/tested in vivo in animal models of CLN3 that are already available to us through our collaborators
Important to the execution of this project and highlighting the role of CLN3 in hiPSC-derived retinal cell model, 1) robust expression of CLN3 protein is observed in hiPSC-derived retinal cells from control (unaffected subjects) (see image below) and 2) JNCL-patient-derived hiPSC-retinal cells display important pathological manifestations of the disease including increased autofluorescent material accumulation and presence of fingerprint inclusions within the cells, the two phenotypic hallmarks of the human disease.
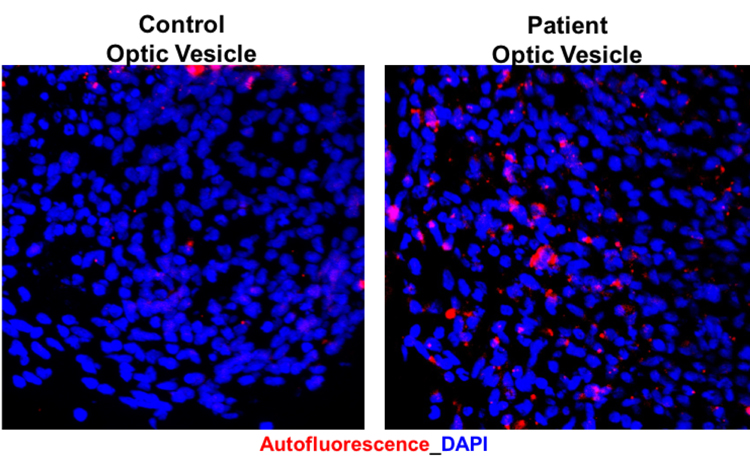
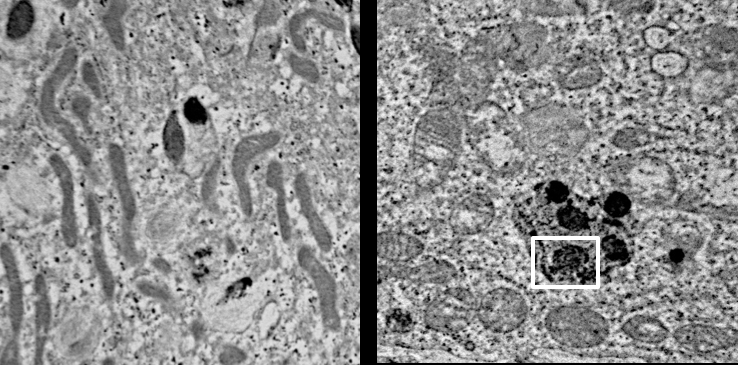
Representative confocal microscopy images showing increased autofluorescence accumulation in JNCL patient-derived hiPSC-optic vesicles compared to control hiPSC-optic vesicle (left) and typical fingerprint inclusions (highlighted within a box) in JNCL patient vs. control hiPSC-retinal pigment epithelium or RPE (right).