Epigenomics
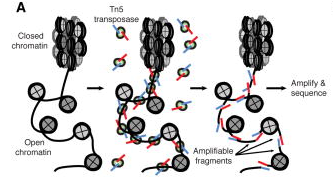
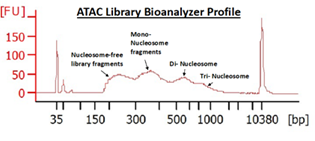
ATAC-seq
ATAC, or Assay for Transposase-Accessible Chromatin (Buenrostro et. al., 2015), utilizes the activity of the Tn5 transposase to insert short segments of DNA into the “accessible” regions of chromatin. These DNA segments are then used to amplify the intervening gDNA to construct a sequencing library. Many studies have correlated these accessible regions with loci of high transcriptional activity, proximal promoter regions of active genes, as well as putative distal enhancer elements.
ATAC-seq input requirements:
- 50,000 freshly isolated cells*
- >90% viability
*Alternative methods allow for snap-frozen cell pellets to be utilized, however fresh cells are preferred.
Following ENCODE guidelines, we typically target 50M sequencing reads per sample.
ChIP-seq
Chromatin Immunoprecipitation can be used to determine the genomic binding sites of your favorite proteins or protein complexes as well as locations of post-translational histone modifications. Typically, ChIP-seq requires millions of cells, treatment with a cross-linking agent to preserve protein-DNA interactions, and fragmentation of the chromatin (usually via sonication) prior to immunoprecipitation with a “ChIP-rated” antibody. Currently, the GRC offers consulting services related to the steps listed above. The GRC maintains a Covaris E220 for investigators to use for chromatin sonication and offers library construction services following immunoprecipitation and DNA isolation.
ChIP-seq& Input Requirements:
- Immunoprecipitations performed in duplicate
- Biological Replicates included
- Input Control (pre-IP fraction) submitted for library construction as well
Following ENCODE guidelines, we target 20M reads for transcription factor IP samples for sequencing. IP samples for broad-peak histone modifications may require up to 50M reads.
CUT&Run
An immediate predecessor of CUT&Tag, CUT&RUN, or Cleavage Under Target & Release Using Nuclease, utilizes protein A-linked MNase in combination with a primary antibody recognizing a Histone post-translational modification (PTM) or transcription factor (TF) in order to identify the genomic sequences associated with the PTM or TF. In contrast to CUT&Tag, the CUT&RUN procedure requires additional steps to prepare the genomic DNA for NGS library construction.
CUT&Tag
Innovated by the Henikoff lab (Kaya-Okur et. Al., 2019), Cleavage Under Targets and Tagmentation utilizes a protein A-conjugated Tn5 enzyme to combine elements of ChIP and ATAC in order to overcome some of the limitations of traditional ChIP-seq. CUT&Tag greatly reduces the number of cells required to perform the assay (from millions to hundreds), eliminates the need for cross-linking and chromatin sonication, and results in sequencing data with better signal-to-noise ratio than traditional ChIP. CUT&Tag is a promising new technology that several University of Rochester labs have recently begun to use with good success. While we currently don’t offer CUT&Tag as a service, we are happy to consult with interested labs and provide guidance.
More detailed CUT&Tag Information:
CUT&Tag General Information
CUT&Tag Protocol
DNA Methylation
Bisulfite conversion of unmethylated cytosines to uracil provides the basis for the following DNA methylation analyses. Amplification of DNA fragments during library construction turns uracil to thymine, which can be sequenced and aligned to the genome using modified alignment parameters. Alignment or mismatch of C/T bases to the genome, allows for decoding the methylated (5-mC, 5-hmC) or unmethylated state of the original DNA sample, with resolution down to single cytosines.
WGBS
Whole Genome Bisulfite Sequencing approaches are employed when the goal is to analyze as many genomic positions as possible, down to single-base resolution.
Following ENCODE guidelines, 30X coverage is suggested in order to statistically define methylation status at single-base resolution.
RRBS
Reduced Representation Bisulfite Sequencing differs from WGBS in that a subset of the genome, enriched for CpG islands, is profiled in the construction and sequencing of the library. MspI digestion of the genome along with size selection of library fragments results in the reduced representation. Since the complexity of the library is substantially reduced, the sequencing depth required for analysis is a fraction of that for WGBS.

Methyl-Specific PCR (MSP)
Methylation-Specific PCR (MSP), can be employed to interrogate specific regions of interest. This process involves designing two sets of PCR primers to the target sequence – one for the native sequence and one for the bisulfite-converted unmethylated sequence. The primer pairs are used in PCR reactions with bisulfite-treated and untreated DNA with gel electrophoresis as a read-out. Quantitative real-time PCR methods are also available. MSP is the most cost-effective approach to query the methylation state of a small to moderate number of specific sequences.
MeDIP
Methylated DNA Immunoprecipitation employs an anti-methylC antibody in order to enrich samples for methylated DNA. Unlike the methods above, there is no need for bisulfite conversion of DNA. Due to the nature of the enrichment, single base resolution is not possible and there is a bias toward hypermethylated regions.

Learn more about Methylation methods from Illumina's Methylation Field Guide.